
ε-HNIW (ε-Hexanitrohexaazaisowurtzitane)
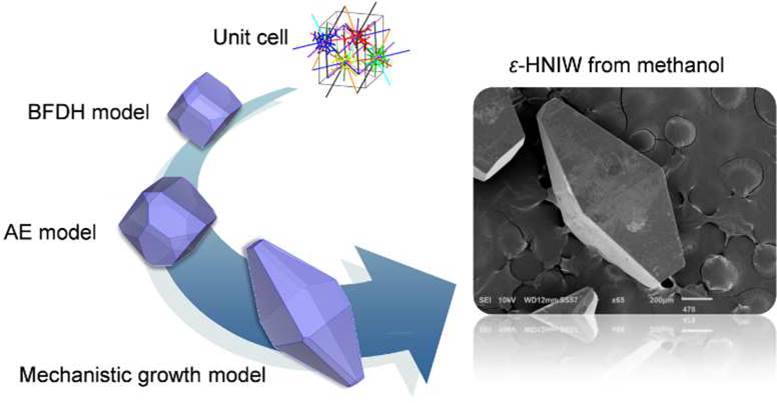
The present work describes how consideration of the onset of supersaturation for 2D nucleation, σ2D, is very important for prediction of the growth habit of ε-hexanitrohexaazaisowurtzitane (ε-HNIW) when the edge energy decreases extremely in solution. From ethyl acetate, the spiral growth model without considering σ2D was shown to accurately predict the polyhedral morphology of ε-HNIW, where the {110}, {101}, {11-1}, {002}, and {10-1} faces are mainly constructed. However, that model was found to be inappropriate for predicting the bipyramidal morphology of ε-HNIW from methanol because the {101} face was anticipated to dominate, but a bipyramid shape is possible only if the {101} face grows faster and finally disappears. The present simulation results show that the edge energy of the {101} face is considerably reduced from methanol by forming a hydrogen bond. This gives rise to a decrease in the σ2D compared with the growth of ε-HNIW from ethyl acetate, which means that the {101} face has a relatively high probability to grow faster by 2D nucleation. As a consequence, a parameter of σ2D enables us to exclude the morphologically unimportant faces among flat-faces by determining which face tends to grow faster by 2D nucleation.
L-Met (L-Methionine)
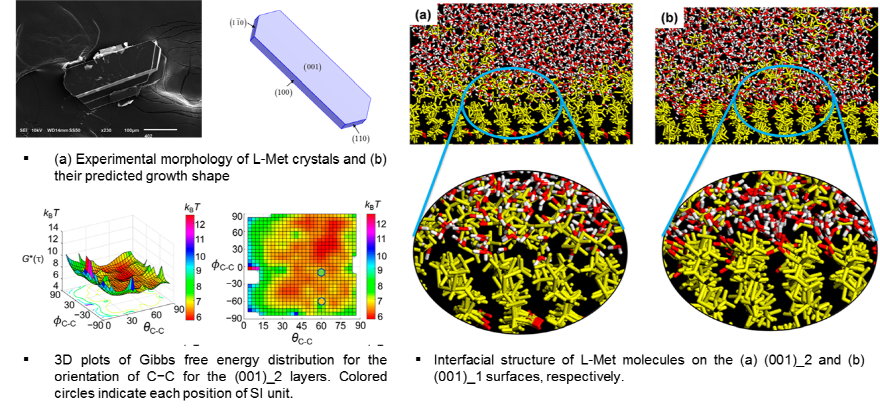
In the present work, the importance of local concentration of the growth of L-methionine (L-Met) was explored by the spiral growth model and interfacial structure (IS) analysis. The spiral growth model shows that the decrease in the local concentration of growth units at crystal faces plays a key role in an appearance of thin hexagonal plate-like morphology of L-Met crystals from water. The (001)_2 layer with hydrogen bonds on its under surface was found to require a relatively large amount of energy for detachment of growth units from kink sites. This situation results in lower local concentration and leads to the decrease in net flux of solute molecules at kink sites compared with the (001)_1 layer interacting with its under surface by van der Waals force. Furthermore, the interfacial structure analysis provides important information on the growth of L-Met. Hydrophilic NH3+ and COO− groups of L-Met on the (001)_1 surface were found to have a tendency to form hydrogen bonding with water, and thus L-Met molecules should overcome the energy barrier required to detach water molecules adsorbed on the crystal surface for continuous growth of the (001) face. From those results, it can be concluded that the limitation of approaching growth units for the formation of the (001)_2 layer onto the (001)_1 surface is responsible for very slow growth of the [001] direction of L-Met crystals.
β-HMX (β-Cyclotetramethylene-tetranitramine)
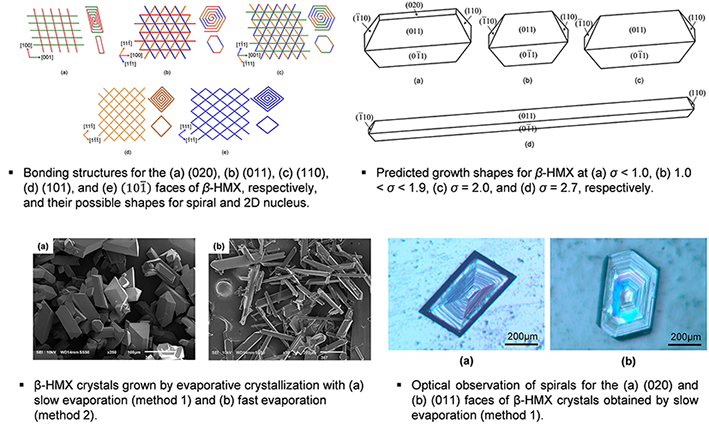
Experimentally, β-cyclotetramethylene-tetranitramine (β-HMX) crystals were found to dramatically elongate to the [100] direction when a relatively high supersaturation was imposed. A sudden growth of β-HMX to the [100] direction is closely associated with a mechanistic transition from spiral growth to two-dimensional (2D) nucleation for the (110) face. The onset supersaturation for the growth by 2D nucleation, σ2D, was found to play a key role in the growth of β-HMX. The present simulation results based on first-principles models such as the spiral growth model and the 2D nucleation model show that the values of σ2D on the (101) and (10-1) faces are smaller than those on the (020), (110), and (011) faces. This leads to the prediction of rapid growth rates for the (101) and (10-1) faces by 2D nucleation at low supersaturation and the appearance of a typical shape of β-HMX. On the other hand, the needle-like shape of β-HMX begins to prevail when the supersaturation exceeds the σ2D for the (110) face because its growth mechanism is transformed from the spiral growth mechanism to the 2D nucleation mechanism which accompanies rapid growth of the (110) face. As a result, the present predictions are in remarkable agreement with the experiments. Furthermore, the kinetic Monte Carlo (KMC) simulation also shows that the σ2D for the (110) face is lower than that for the (011) face because the (011) face provides the surface topology on which growth units are unfavorably incorporated into the lattice sites. It evidently shows that the relative positions of σ2D bring on the advent of needle-like growth of β-HMX.
DADNE (1,1-Diamino-2,2-dinitroethylene)
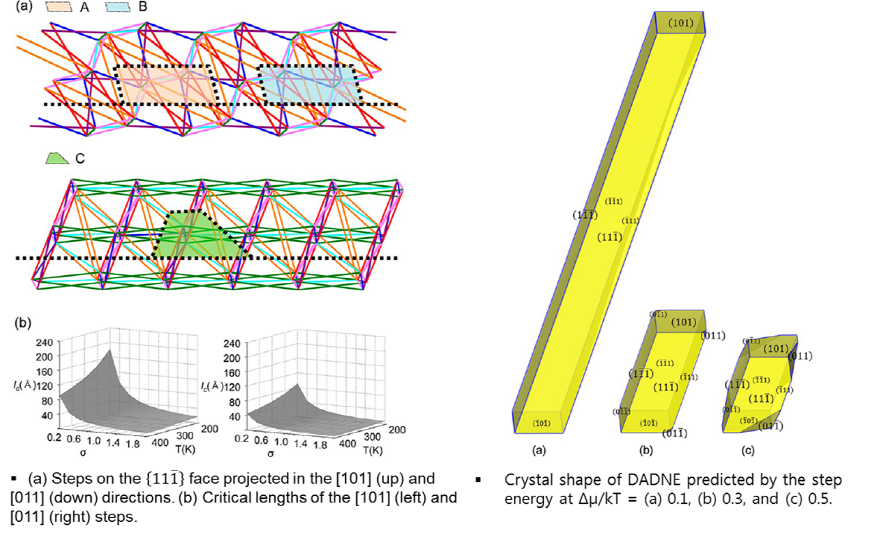
In the cooling crystallization of 1,1-diamino-2,2-dinitroethylene (DADNE), it was found that the aspect ratio of crystals decreases as the cooling rate of the solution increases. To reveal the effect of supersaturation on the growth shape of DADNE, molecular modeling was carried out by the step energy calculation and kinetic Monte Carlo (KMC) simulation. The rodlike shape of DADNE with basal {11-1} faces was accurately predicted by the step energy calculation, resulting in remarkable agreement with the experiments. The reason behind the slowest growth rate of the {11-1} faces was found to originate from the high energy barrier in the formation of a 2D nucleus on the crystal face. Furthermore, it was shown that the aspect ratio of DADNE decreases by the lowered free energy of 2D nucleation at high supersaturation, in which the distinctive characteristics on the anisotropic growth behavior of DADNE are blurred. The KMC simulation results also provided an understanding of the growth kinetics of growth units on each crystal face: the {11-1} face shows a lower sticking fraction, which means that the {11-1} face offers the surface topology where growth units are difficult to incorporate into the lattice sites. However, as the supersaturation increases, the crystal faces start to be strongly roughened, and the aspect ratio becomes reduced.
AP (Ammonium Perchlorate)
The simulation system that yields reliable physicochemical propertiesof ammonium perchlorate (AP) such as lattice energy, solution density, diffusion coefficient, and radial distribution functions was constructed. Dissolution of AP in water was computationally confirmed to be an endothermic reaction, which corresponds to the fact that crystallization of AP from an aqueous solution is an enthalpically favored process. On the basis of the Yasuoka−Matsumoto method, the MD simulation on the homogeneous nucleation of AP from an aqueous solution was carried out, and the calculated nucleation rate was shown to be 1 order of magnitude different from that predicted by the classical nucleation theory (CNT). The critical sizes of nucleus (12 and 8 at S = 5.1 and 6.4, respectively) are also very close to those estimated by the CNT (12.4 and 8.2 at S = 5.1 and 6.4, respectively).
RDX (Hexahydro-1,3,5-trinitro-1,3,5-triazine)
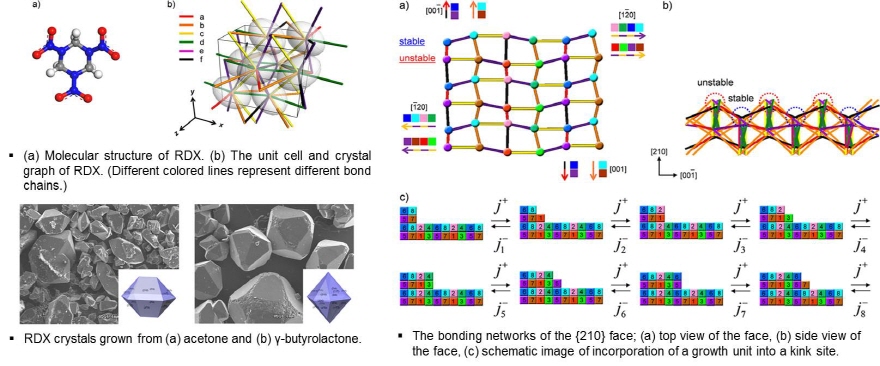
The crystal morphology of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) was predicted by the advanced Burton−Cabrera−Frank (BCF) model with consideration of non-centrosymmetric growth units. The present modeling showed that the advanced BCF model provides reliable results and understanding of the growth habit of RDX crystals grown from acetone and γ-butyrolactone, in which the {210} and {111} faces are dominantly developed.
AS (Ammonium Sulfate)
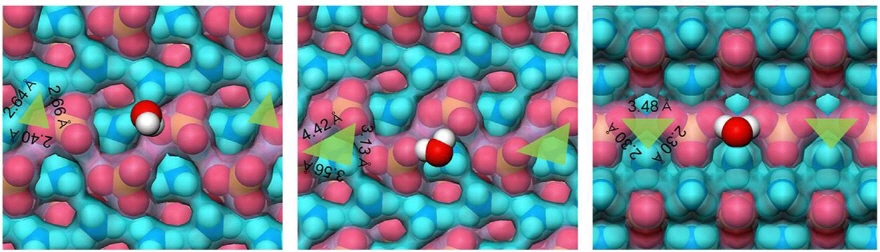
Growth mechanism of the (100) and (001) faces of ammonium sulfate was found to be different even at the same supersaturation. The growth rate of the (100) face was mostly proportional to the solutionsuper-saturation.On the other hand, the (001) face began to grow after reaching a reasonably high supersaturation.The delay in growth of the (001) face at low super saturation inferred to the strong interaction between crystal surface and solvent molecules was explained by molecular modeling. The calculated binding energy of a water molecule with each faces showed that the water molecule is mores trongly adsorbed on the (001) face than the(100) face.The symmetrical arrangement of ammonium sulfate molecules was shown to provide a good binding site in which a water molecule is more strongly adsorbed on the crystal surface and the relative affinity to the (001) face is also larger than that of the (100) face.Those calculation results strongly support the growth mechanism of the (001) face at low supersaturation.
γ-Glycine
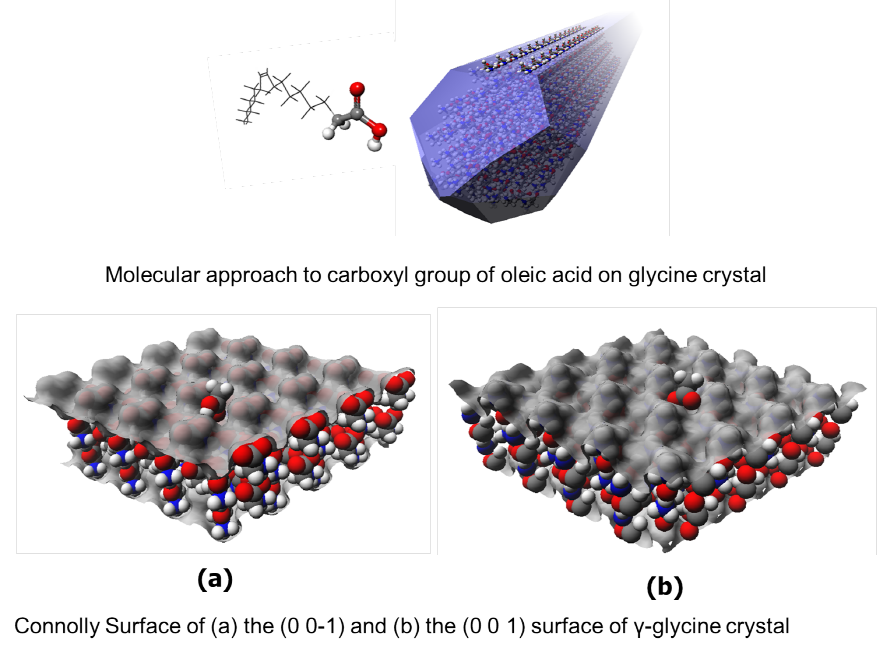
In general, α-glycine is thermodynamically most stable form in water. However, γ-glycine was found to be mostly produced from the emulsion in which water droplets were dispersed in oleic acid. The mechanism on the polymorphism of lycine in the particular enviroment was studied by molecular modeling.
ADNBF(7-Amino-4,6-Dinitrobenzofuroxan)
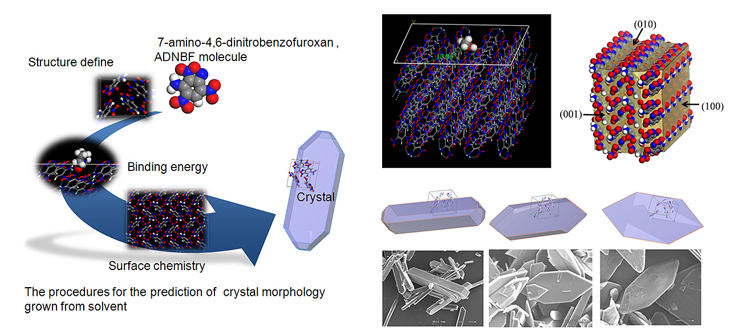
It has been known that solvents, additives, or impurities alter the growth rate of certain crystal faces during crystal growth by their strong adsorption on specific faces of the crystal. The calculation of solvent−surface interaction energy based on molecular dynamics has been suggested as a way to predict the solvent-mediated growth habit.